药品不良反应核实报告范文 GMP文件的编制管理内容(3)
记录必须与标准保持一致
可将最终标准要求列入记录中
文件编制与管理过程
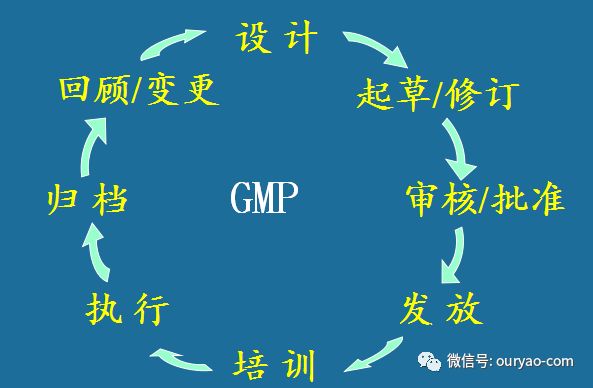
文件的设计
明确组织机构体系中相关职责部门的职责
成立跨职能工作小组(CFT)
安排与提供适宜的,有效的培训
对原有文件作系统的回顾与评价 *
依据现行GMP的要求,作出改进计划*
文件的编码
系统性:统一分类、编码
准确性:文件应与编码一一对应
可追踪性:可随时查询文件的变更历史
稳定性: 应保证系统的稳定性
相关一致性:文件一旦变更,相关文件中出现的该文件号同时进行修正

文件的起草/ 修订
起草/修订:由文件主要使用部门负责
“ 缺什么,补什么”
“边执行,边修订,边完善”
视作为GMP培训与提高的机会
起草文件的基本要求:
文件标题、类型、目的、适用范围应清楚的陈述
用词简洁、准确
流程清晰、职责分明
文件如需记录,应有足够空间
附上必要的流程图及记录样本
应表明文件编码、总页数及分页号

文件的审核/批准
所有文件的审核人与批准人必须预先规定
审批人具有相应的资格与能力
所有正式生效的文件,均应有起草、审核、批准人签字,并注明日期

文件的发放
文件一旦批准,应在执行之日前发放至相关人员或部门
文件发放必须进行记录
新文件执行之日必须收回过时的文件
文件的培训
新文件必须在执行之日前进行培训并记录
培训师原则上为文件的起草者、审核者或批准者
必须保证文件使用者均受到培训
文件的培训是GMP培训的重要组成部分
培训的方式为:自学/培训课、提问与回答、模拟演练、实地一对一授教等
所有培训均应记录
文件的执行
文件的有效执行是最重要的环节
新文件初始执行阶段,应特别注意监督检查执行情况
所有文件必须定期进行复核
应定期向使用部门提供现行文件清单
对现行的管理标准,未经变更控制,不得随意改动文字内容
对现行的批记录或其他记录文件,确有必要进行改动,应由责任人签字,并注明原因和日期
采用自动控制或管理系统记录,应仅允许授权人操作
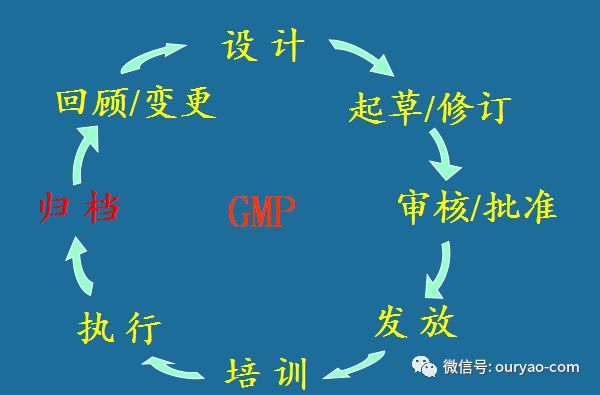
文件的归档
所有的现行文件原件或样本均存档于质量管理部门
所有的过时文件仅保留一份在质量管理部门
记录按种类归档,按其重要性分别保存在相关部门并存档至规定日期
各种归档文件应建立台帐以便调用
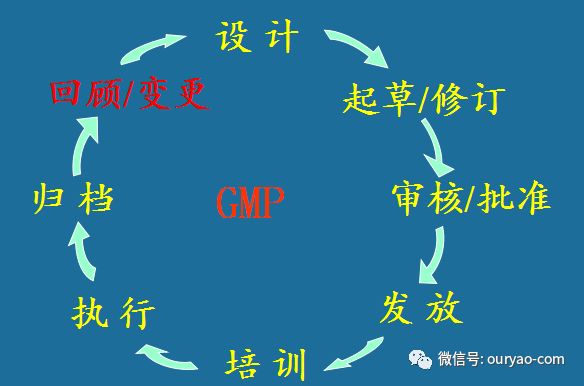
文件的回顾
应定期对重要的管理标准作相应的回顾
文件的变更控制
任何文件未经批准不得随意更改
变更的提出:文件的使用者或管理人员有权提出变更
变更的审批:应评价变更的可行性并批准变更,履行变更手续
变更的执行:变更执行过程可视为一份新文件起草
世界警察&