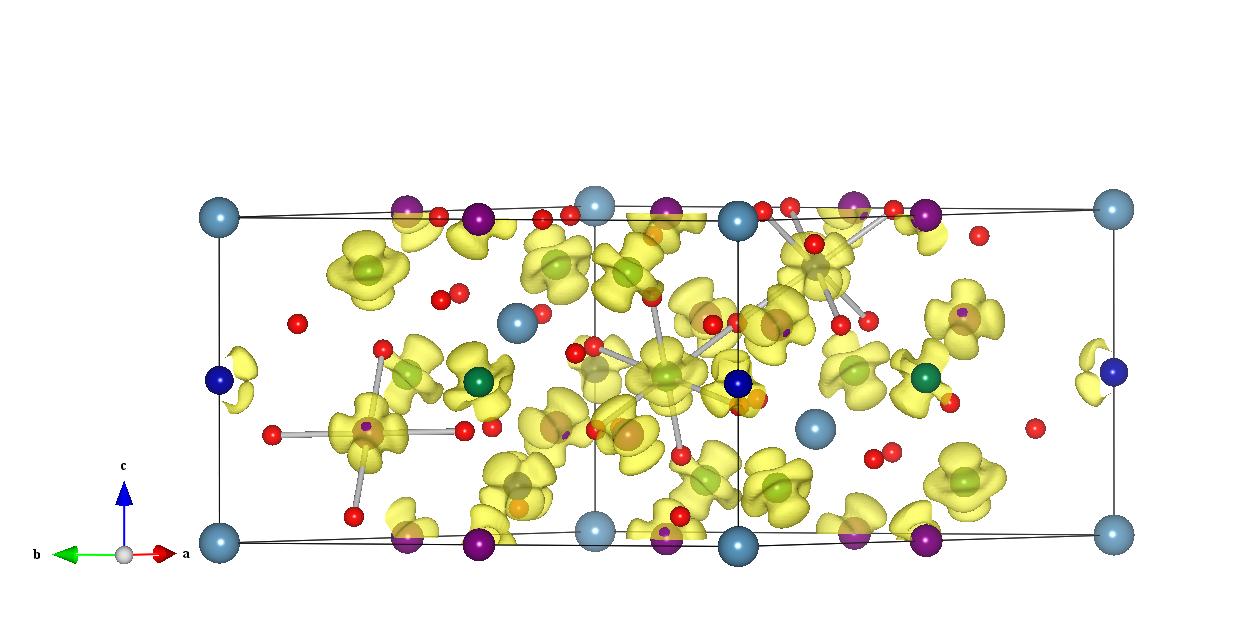
差分电荷密度CDD
Sample Text
大家好以前我分析成键都用DOS+ELF+Bader 最近的一个工作我师姐建议做下电荷差分密度(charge density difference)我没做过请大家帮忙谢了先!
1)求用vasp做过电荷密度差分的文献这个用ISI实在不好查老板催工作我时间又不多--如果同学手上有现成的文献给我参考感激不尽
2)查了下论坛原来的帖子找到一个我认为比较好的参考贴(我不知道如何给他金币,版主看到了教下我,呵呵),渔火江枫的先引用下:
VASP与电荷密度差
一般分为:差分电荷密度图(deformation charge density),二次差分电荷密度图(difference charge density)
两者的区别有很多种说法,其中一种定义认为:
差分电荷定义为成键后的电荷密度与对应的点的原子电荷密度之差。通过差分电荷密度的计算和分析,可以清楚地得到在成键和成键电子耦合过程中的电荷移动以及成键极化方向等性质。
“二次”是指同一个体系化学成分或者几何构型改变之后电荷的重新分布。
如何做对电荷密度做差,目前有两个版本的公式:

人认为公式(1)和差分电荷密度的定义比较吻合,但是这种方法会产生很多干扰信息,而文献中常用的是公式(2)。
如何用VASP做电荷密度差图?
①对优化后的结构做静态自洽计算
同一个应用,相同的图片分别放在drawable-xxhdpi、drawable-xhdpi、drawable-hdpi、drawable-mdpi、drawable-ldpi中,在同一设备中占用的内存会大不一样(设备的dpi是固定的,图片放在不同的dpi文件夹下,在设备上显示时需要将图片转换成和当前屏幕一样dpi后在设备中显示,所以即使该图片在不同dpi文件夹下大小一样,但放在内存中的大小却不是一样的,并不一定是长*宽*4),做应用的内存优化之前可以先看一看你的工程是如何做屏幕适配的,是否有优化的空间。【知识点的认识】帕斯卡定律:帕斯卡大小不变地由液体向各个方向传递.大小根据静压力基本方程(p=p0+ρgh),盛放在密闭容器内的液体,其外加压强p0发生变化时,只要液体仍保持其原来的静止状态不变,液体中任一点的压强均将发生同样大小的变化. 这就是说,在密闭容器内,施加于静止液体上的压强将以等值同时传到各点.这就是帕斯卡原理,或称静压传递原理.应用:万吨水压机、千斤顶,液压机等.。说明要有顺序,这是使说明内容条理化的必要条件.常见的说明顺序有:时间顺序、空间顺序、逻辑顺序.说明的时间顺序和记叙的时间顺序相似.空间顺序,要特别注意弄清空间的位置,注意事物的表里、大小、上下、前后、左右、东南西北等的位置和方向.逻辑顺序,常以推理过程来表现.采用什么顺序,主要取决于作者所说明对象的特点.说明事物的发展变化,时间顺序容易表示清楚.写建筑物的结构,离开空间顺序难让读者看明白.说明事理用逻辑顺序,正便于体现事理的内部联系.。
INCAR中几个注意的参数:
IBRION =-1;NSW= 0;
NGXF,NGYF,NGZF
②做差
将第一步得到的三个CHGCAR文件依次命名为CHGCAR1、CHGCAR2、CHGCAR3,采用脚本程序按照公式②对三个文件做差,得到CHGdiff文件。
③视图
3D显示可以将得到的CHGdiff文件添加vasp后缀(CHGdiff.vasp),用VESTA读取,另外VESTA也可以做2D切面,缺点是没有标度。
2D的显示也可以用lev00去做切面,然后用origin作图,得到的2D面电荷密度差带有标度便于分析(具体做法可参阅lev00说明书)。
好了我的问题就是比如我的体系基底是A材料然后呢是多个B原子吸附或者是嵌入(这里你视为化学键合的作用就行),形成了A(B)n,对上面的公式2应用到我的体系如何处理?比如说步骤中的1,2步 B做静态自洽计算的时候是n个B一起算(我理解是扣除A的所有坐标得n个B的坐标,不知对否)还是一个一个B分开算,如果是分开,那脚本做CHGdiff如何做呢?如果都不是,请教下我该如何做,谢谢!
举报删除此信息
广告
cavediger(站内联系TA)
: originally posted by adan0553 at 2011-09-19 10:07:17:。originally posted by fengqwen1314 at 2011-04-17 10:30:14:。originally posted by tlltll at 2011-04-10 17:41:16:。
Sample Text
大家好以前我分析成键都用DOS+ELF+Bader 最近的一个工作我师姐建议做下电荷差分密度(charge density difference)我没做过请大家帮忙谢了先!
1)求用vasp做过电荷密度...
差无定法,怎么差要看你要什么
xiao72379(站内联系TA)
originally posted by kingoywm at 2011-05-30 19:48:36:。originally posted by newdings at 2011-01-19 23:50:05:。originally posted by sujingzuo at 2011-05-19 20:32:15:。
差无定法,怎么差要看你要什么
对同一元素的原子,由于核电荷不变,失去的电子越多,有效核电荷增加得越多,核对外层电子的吸引力越大,剩下的电子就越不容易失去,所以i1 cavediger(站内联系TA) originally posted by kingoywm at 2011-05-18 18:45:19:。originally posted by sujingzuo at 2011-05-19 20:32:15:。originally posted by newdings at 2011-01-19 23:50:05:。 要这些原子也就是上面说的B与体系A的成键信息或者说看电荷转移目的还是挺明确的我现在是不知道怎么操作以前没做过现在组里也没人教具体疑问上面已经提出来了如是说算一个B+A---AB我按帖子应该做的出来... 如果B成团簇,讨论整个团簇与A相互作用,那就把B一起算;如果不是,B比较弥散,且周围环境等同,彼此分开较远,也可以一起算;如果B周围环境不等同,B、A作用也不同,那就一个一个地算;如果考虑B吸附或嵌入后对其他B的影响,先吸附/嵌入上去的B要同A一起作基底,其它B单独算...... 漂泊四方(站内联系TA) Originally posted by xiao72379 at 2011-05-25 10:17:31: Sample Text 大家好以前我分析成键都用DOS+ELF+Bader 最近的一个工作我师姐建议做下电荷差分密度(charge density difference)我没做过请大家帮忙谢了先! 1)求用vasp做过电荷密度... 好了我的问题就是比如我的体系基底是A材料然后呢是多个B原子吸附或者是嵌入(这里你视为化学键合的作用就行),形成了A(B)n,对上面的公式2应用到我的体系如何处理?比如说步骤中的1,2 步 B做静态自洽计算的时候是n个B一起算(我理解是扣除A的所有坐标得n个B的坐标,不知对否)还是一个一个B分开算,如果是分开,那脚本做CHGdiff如何做呢?如果都不是,请教下我该如何做,谢谢! 方案一里面:把B当做一个整体,主要考虑的是A整体和B的作用。 方案二里面:实际上考虑了A和B的作用,还考虑了B与B之间的作用。 所以重点看你关注什么,衬底与吸附原子之间的作用还是吸附原子之间的作用。两种作用之间的强弱关系?看你的意思。电荷密度 定义如果B形成了团簇,方案一可能是你关注的,如果B比较离散,作用力较弱,方案一也可以。如果BB之间作用力介于他们之间,和衬底的作用类似,分析起来就比较吃力了。 操作起来其实还不是太难,记得p4vasp好像可以做这个。 xiao72379(站内联系TA) Originally posted by 漂泊四方 at 2011-05-26 08:43:26: 化学吸附:吸附质分子与固体表面原子(或分子)发生电子的转移、交换或共有,形成吸附化学键的吸附。1.疏水参数计算参考值:无2.氢键供体数量:03.氢键受体数量:74.可旋转化学键数量:185.互变异构体数量:无6.拓扑分子极性表面积1187.重原子数量:298.表面电荷:09.复杂度:54610.同位素原子数量:011.确定原子立构中心数量:012.不确定原子立构中心数量:313.确定化学键立构中心数量:014.不确定化学键立构中心数量:015.共价键单元数量:2。1.疏水参数计算参考值:无2.氢键供体数量:03.氢键受体数量:44.可旋转化学键数量:95.互变异构体数量:无6.拓扑分子极性表面积36.97.重原子数量:128.表面电荷:09.复杂度:67.510.同位素原子数量:011.确定原子立构中心数量:012.不确定原子立构中心数量:013.确定化学键立构中心数量:014.不确定化学键立构中心数量:015.共价键单元数量:1。 我的情况是:B在体系A表面不成团簇,B与A体系的作用从bader计算看是有一个|e|左右的电子从B转移到体系A(B是2价主族元素) DOS上看B的S电子与体系A的峰有杂化现象,呈现一定的共价成分,不过B的S电子在费米能级附近有明显裂分现象,一个峰出现在价带,一个峰出现在导带,还是很好的印证了bader电荷分析的结果。所以我初步判断了B与A体系的键合作用是主离子性辅共价性的混合键性。做电荷差分是进一步支持下我的论点。我在计算过程中除了考虑一个B与A的作用,也做了多个B与A的作用。当然一个B与A的作用做电荷差分现在看来我在步骤上已经是清楚了,但当多个的时候,情况应该是A与B是较强的化学作用,BB之间作用较弱,从ELF做的图看,没有化学键,顶多是静电排斥,我主要关心的是B与A的键合情况是不是我上面提到的情况,这个差分做出来也是为了多一个证据说明。写的有点乱,总结下就是:B不成团簇,B较分散,但不同B的化学环境是不同的,因为他们嵌入的位置不一样,不过成键性质是一样的(比如dos+ELF的结果显示定性是类似的,不过是定量上的不一样)。我确定B与A之间有化学键合,我希望做差分进一步研究成键情况,当然结果能匹配我上面提到的我已经做过的一些成键分析结果是最好的了。如果是B一起做个差分(考虑到相同键性),我还能分析到每个B与A的电荷转移情况?还是该一个一个来?如你做过,能不能给个细致点的操作步骤,如何处理vasp文件进行计算得到差分结果? xiao72379(站内联系TA) Originally posted by cavediger at 2011-05-26 05:43:58: 如果B成团簇,讨论整个团簇与A相互作用,那就把B一起算;如果不是,B比较弥散,且周围环境等同,彼此分开较远,也可以一起算;如果B周围环境不等同,B、A作用也不同,那就一个一个地算;如果考虑B吸附或嵌入后... cavediger(站内联系TA) Originally posted by xiao72379 at 2011-05-26 09:56:52: 到了工作时候吧 朋友介绍我玩dh3 记得还是07年11月底吧 那时候好像不要什么点卡我们就玩了2个月吧觉得没多大意思,就很少玩有一个月玩那么几次到了08年9月左右吧 才95级还没转.. 就没玩了,杭州打折机票,后来08年快过去了就打算开店吧.想想09年要是开了店.在店里无聊怎么过啊.在家又把dh3下载了 弄了个家族人还蛮多200多个 .新区一开我们家族就进去了 我弄了一个帮派里面全是家族里的人.以前玩游戏从来没想过结什么婚,上海到南昌机票,弄了家族成了帮主后家族里人老叫我去找个结婚,我就随便找了一个结婚.那次还真搞笑结婚都不知道怎么弄.弄得那女的哭笑不得.结婚不到5天吧就离了.晕是我太小了,我汗了我还没嫌弃她26呢.搞笑.后来吧我80几级,广州到深圳机票,偶然看见世界有个叫宅女的在找老公.我就m了她让她嫁我.汗. 她居然第一句回我,就是你养我啊.当时觉得只是游戏嘛好玩而已就说了当然养你啊. 后来她加了我那个家族群.结婚之前记得帮她买了把仙器,过了一段时间我们就在游戏里结了婚.她双开没点卡了呢就跟我说我就帮她冲,要是没冲就很生气.。就是这个时候,看到进入了最佳阻击位置,一号立刻就点了屏幕上的开关,借着李从就看见那个废桶子一样的卫星立刻就改变的自己的形象,变成了一个气象卫星模式的样子,而且接着就在它中间的腹部位置伸出了一个类似于长枪的东西,当然李从可不会傻的以为那就是一杆长枪的,那肯定就是攻击武器了,这隐藏的还真是比较严实呢,没想到一个外表废物的东西其实还是一个“变形金刚”呢,要是美国航天局的人监测到了,会不会觉得这个东西是从赛博坦星球来的呢。另一个刚才跟他爸出去了,剩余的一个空荡荡的床铺没有人来,反正也没有什么事,就一个人在nh的校园里逛一逛吧,那时候,双脚的水泡疼得要命,第一次来大学,真心感觉大学为什么会这么大的,(我觉得所有有这个想法的学生,一定会在两三个星期之后,当他们真正逛完这个大学的时候就会发现,怎么我上的大学这么小,别人家的大学校园怎么这么大,为什么我们大学的校园这么破而且难看,连一个约会的地方也没有之类的话,哈哈,好像没一个中国学生都会对自己的学校不满足)一个人在校园里逛着,看着身边经过的一个个学姐,只是觉得他们比高中时候的女生更会打扮了,但是却没了那几份清纯,反正,我也对你们不感兴趣。 漂泊四方(站内联系TA) Originally posted by xiao72379 at 2011-05-26 09:56:52: 其实做计算的很多情况下都不是做之前就能了解结果的。对于你这个问题最简单的办法就是都去做一遍,然后看看那个结果更符合你的要求,因为感觉上判断这两个方法对你的体系来说没有太大的区别。电荷密度 定义只有几个原子的体系,计算应该不会花很多时间吧? gloomy2004(站内联系TA) 很有收获,谢谢! gloomy2004(站内联系TA) 有一个想法,我打不出密度那个字母,用p代替,请勿介意。 e是这五种元素中原子半径最小的元素,则它们的原子序数由小到大的顺序是( )a.a、b、c、d、e b.e、c、d、b、a c.b、a、d、c、e d.c、d、a、b、e5.下列各组顺序的排列不正确的是( )a.离子半径:f->na+>mg2+>al3+ b.热稳定性:hcl>h2s>ph3>ash3c.酸性强弱:h3alo3<h2sio3<h2co3<h3po4 d.溶点:金刚石>na>sio2>co26.某元素原子的质量数为a,它的阴离子xn-核外有x个电子,w克这种元素的原子核内中子数为( )a. b.c. d.7.某主族元素r的最高正价与最低负化合价的代数和为4,由此可以判断( )a.r一定是第四周期元素 b.r一定是Ⅳa族元素c.r的气态氢化物比同周期其他元素气态氢化物稳定d.r气态氢化物化学式为h2r8. 元素x和元素y在周期表中位于相邻的两个周期,x和y两原子核外电子总数之和为19,y原子核内质子数比x多3个,下列叙述正确的是( )a.x和y都是性质活泼的元素,在自然界中只能以化合态存在b.x和y形成的化合物的化学式为y2xc. y的化合物种类比x的化合物种类多d.y能置换酸中氢,放出氢气,但不能置换出盐中的金属9. 运用元素周期律分析下面的推断,其中不正确的是( )。(2)中b原子最外层三个电子形成三条共价键,最外层共6个电子,h原子达到2电子稳定结构,只有氧原子形成两条键达到8电子稳定。丁克毅,等[9]将亲水性组分连接在pa上,形成以pu为核,pa为壳的核/壳结构,这种结构的乳液,在丙烯酸酯分子和聚氨酯分子之间不存在较强的、直接的化学键合作用,通过聚氨酯对丙烯酸酯分子的包覆作用,羧基产生的电荷稳定作用以及氢键和分子间力作用,使得体系热力学稳定。 另外, LZ可不可以把那个相减的脚本发给我?我不太会写那玩意。谢谢。happygloomy2004@yahoo.com.cn gloomy2004(站内联系TA) 我的意思是,一个个a,b往进加。 阿黛拉(站内联系TA) : Originally posted by xiao72379 at 2011-05-25 10:17:31: Sample Text 大家好以前我分析成键都用DOS+ELF+Bader 最近的一个工作我师姐建议做下电荷差分密度(charge density difference)我没做过请大家帮忙谢了先! 1)求用vasp做过电荷密度... 6 你好 早上好 ,请问, 谢谢,。最重要的四句话:你好请问谢谢没问题。你好,请问可以给我发送一份江小媚作品么,谢谢,471400662@qq.com xiaolongha...:亲,你好、 你要的文件已经发送, 如果未收到、请稍等片刻, 若有关于此问题的疑问处、 可以选择追问我...。
20