药品不良反应制度 浅谈日本药品审批审评制度和市场准入机制(2)
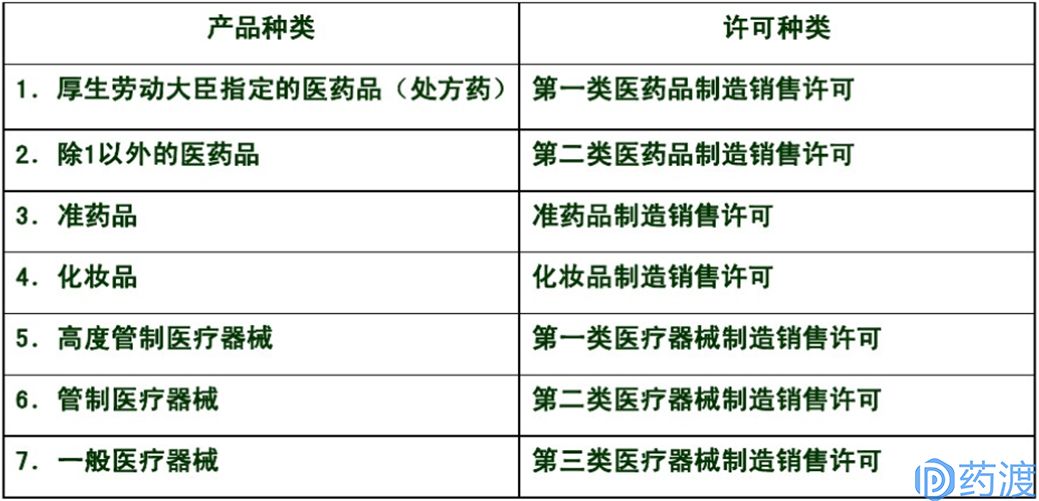
图2.日本制造销售业许可类型
②制造业许可
对于日本国内药品制造企业,《药事法》第十三条规定:除非药品、准药品或化妆品的生产厂商获得制造业许可,否则每个人不得制造药品、准药品或化妆品。制造业许可五年一更新。所有想要获得制造业许可的企业其生产设备和厂房建筑必须要符合药房建筑设备条例等相关法规要求,并根据需要生产的药品选择相应的制造业许可。制造业许可共有以下五种类型:生物制品许可,放射性制品许可,无菌制品许可,普通制品许可和分包装、标签、贮存许可。
对于日本国外的药品制造企业,《药事法》第十三条之三规定:有意制造出口到日本的药品、准药品或化妆品的人(以下简称“外国制造商”),可以由厚生劳动大臣认定,以取得国外制造厂商认定。国外制造厂商认定的有效期为5年,每5年需要进行更新。国外制造厂商认定所需要的厂房和设备条件与制造业认可完全一致。
③制造销售业认可
对于想要在日本上市的每一种药品,《药事法》第十四条规定:有意制造和销售含有厚生劳动大臣指定成分的药品、医药外用品和化妆品的人,必须为其制造和销售每个产品得到厚生劳动大臣的认可。而获得制造销售业认可的必要条件为申请人获得制造销售业许可,产品制造厂商获得制造业许可(或国外制造厂商认定)。有意取得制造销售业认可的申请人应根据厚生劳动省条例的规定,将与临床试验结果相关的文件和其他材料附加在申请书上。在这种情况下,当与上述申请有关的药品是厚生劳动省条例规定的药品(即处方药)时,应根据厚生劳动省条例规定的标准收集和准备材料。
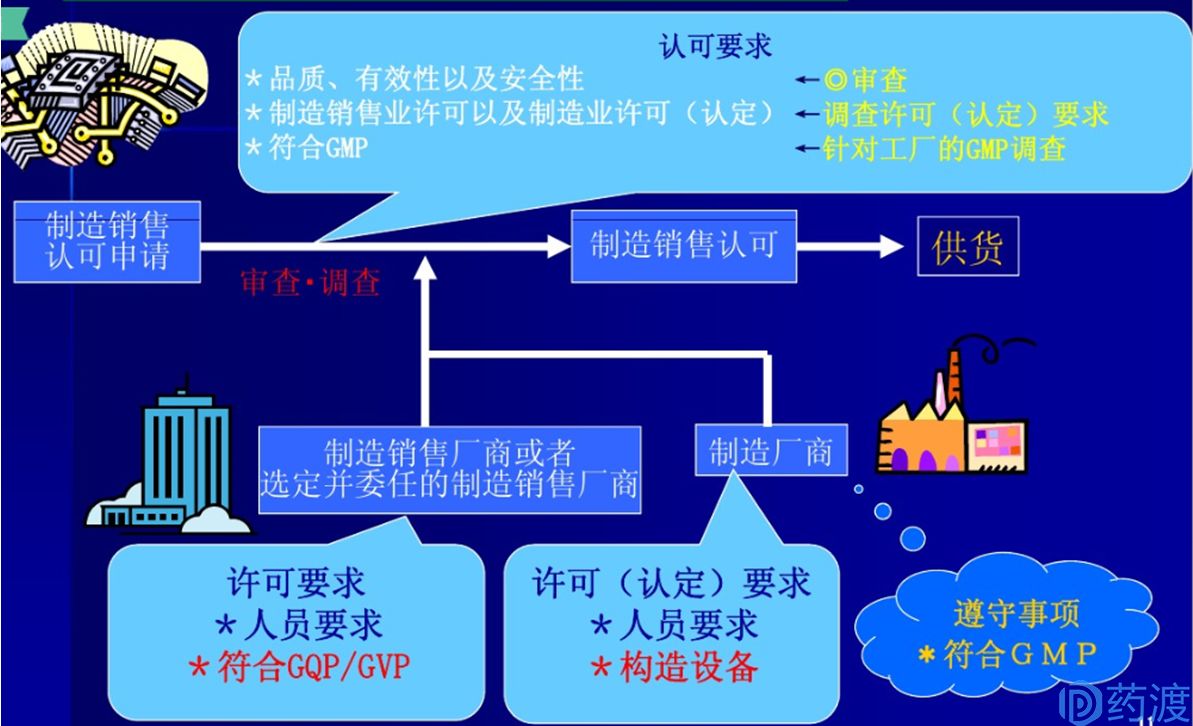
图3.日本医药品制造销售认可许可制度
药品制造销售业认可流程

为了保证在日本上市的药品的有效性、安全性和质量可控性,每一个上市药品都需要获得制造销售认可(由厚生省颁发),制造销售认可申请的审评由PMDA完成。日本药物注册分为以下10类,仿制药归属于第10类。

在日本申请上市的申报资料材料如下表所示:

不同注册分类的药物需要的申报资料不同,具体如下图所示:
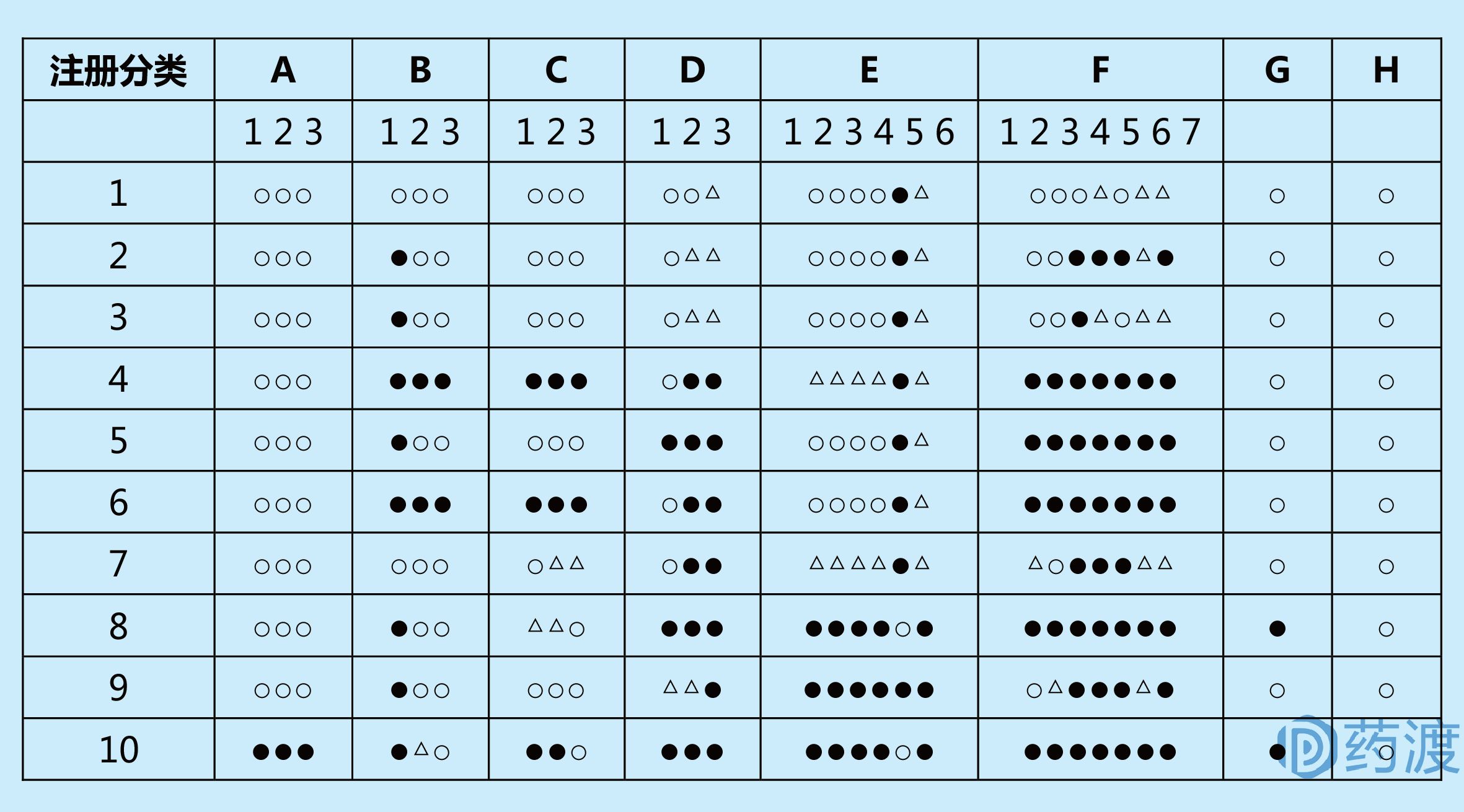
注:○必须要求的材料;● 不要求的材料;△ 视情况而定的材料
日本新药申请从2003年开始采用ICH规定的CTD格式,仿制药申请从2017年开始采用CTD格式。对于优先审评审批的产品,审评时间在9个月左右,普通申请的审评时间在12个月左右。国外药物生产企业可以直接申请产品的制造销售认可,在这种情况下,国外药物生产企业需要在日本国内具有销售制造许可的企业中选定一家指定其为MAH,该日本MAH承担相应的在日本上市产品的安全、有效和质量可控责任。
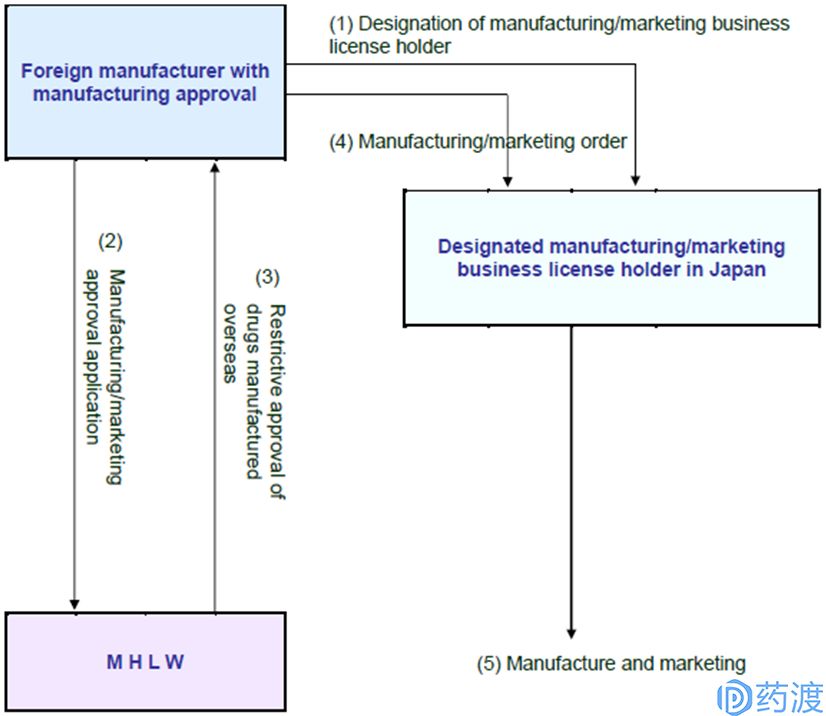
图4.国外药物生产企业在日本申请销售制造认可流程图
小结
我国药企在将国产药品出口到日本市场的过程中首先要取得国外制造厂商认定,其次必须要与日本当地企业进行合作,因为日本监管法律不允许其他国家企业作为产品MAH存在。药品不良反应制度在这种情况下,日本市场的准入门槛更高,能够顺利在日本上市的国产产品从一个方面证明了企业生产过程的规范和产品质量优秀。目前CFDA对于欧美日共线品种表示认可,也可见我国在加入ICH以后积极与其他成员国实现品质、监管互认,也一定程度上推动了国产药品迈步走出去。
参考文献
[1]日本2017年版《药事法》
-

-
师冠超
思想退化到了腐男的地步
-
陈云
那就是解放军雄赳赳气昂昂
-
谭峭
给我一次机会我想刷回9
-
自己没理由把伊拉克打碎了还在干协别人站队